

CO TO JEST FENYLOKETONURIA?
Fenyloketonuria jest wrodzoną chorobą metaboliczną występującą z częstością około 1 na 8 tys. noworodków (ang. : PHENYLOKETONURIA w skrócie PKU).
U podłoża choroby leży nieprawidłowość przemiany w organizmie chorego jednego z elementarnych składników białka, aminokwasu – fenyloalaniny. O ile odpowiednio wcześnie nie zostanie zastosowane właściwe leczenie, gromadząca się w nadmiarze fenyloalanina uszkadza organizm dziecka, a w szczególności jego centralny układ nerwowy (mózg!), prowadząc nieuchronnie do upośledzenia umysłowego. Stąd konieczność wczesnej diagnostyki choroby opartej o badania przesiewowe noworodków i wczesnego prowadzenia leczenia dietą o ograniczonej podaży fenyloalaniny. Takie postępowanie warunkuje profilaktykę uszkodzenia układu nerwowego pozwalając na całkowicie prawidłowy rozwój dziecka chorego na fenyloketonurię.
JAK DZIEDZICZYMY PKU
Fenyloketonuria dziedziczona jest drogą przenoszenia nieprawidłowości w przemianie fenyloalaniny (defekt enzymu) w sposób „ustepujący” (tzw. Recesywny) przez oboje rodziców. A zatem choroba może wystąpić (ale nie musi) u dziecka jedynie wtedy, gdy oboje rodzice są nosicielami „wadliwego” genu. Nosiciele choroby sami są zdrowi. Wśród potomstwa dwojga rodziców nosicieli, ¼ dzieci może być zdrowych, ½ może być zdrowych, lecz będą nosicielami choroby, a ¼ dzieci może być chorych na fenyloketonurię.
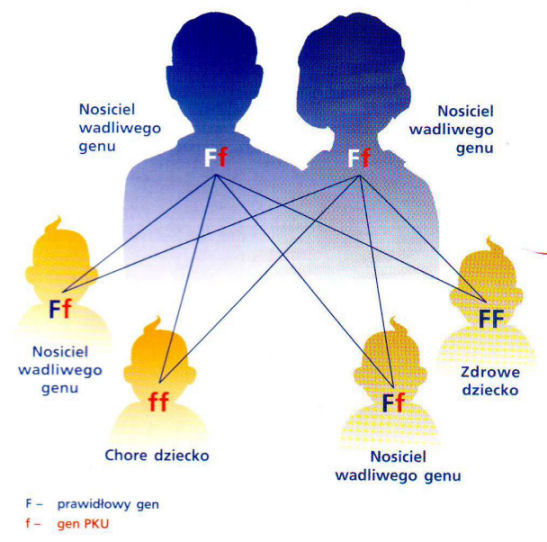
Ryzyko wystąpienia fenyloketonurii u potomstwa nosicieli choroby wynosi 25% (1 na czworo dzieci), ale w praktyce może jednak zdarzyć się wystąpienie choroby u dwojga, a nawet trojga dzieci w rodzinie. Przy każdej ciąży ryzyko choroby wynosi 25% i nie istnieje gwarancja, że po urodzeniu chorego, kolejne dzieci będą zdrowe.
Zatem fenyloketonurią nie można się zarazić ani nabyć jej wyłącznie od jednego z rodziców.
Chore dziecko może urodzić się tylko wtedy, gdy nosicielami są oboje rodzice. W razie nosicielstwa jednego z rodziców dzieci będą albo zupełnie zdrowe, albo będą to zdrowi nosiciele, ale nigdy osoby chore.
PKU – TROCHĘ HISTORII
dr Robert Guthrie (1916 – 1995)
Dr. Guthrie wynalazł stosowany dziś powszechnie tzw. screening test. Test, dzięki któremu poprzez pobranie próbki krwi, można wykryć fenyloketonurię. Szacuje się, że uratował on ok 30000 osób przed ciężkimi skutkami tej choroby. Dr Robert Guthrie zajął się badaniami w tym kierunku z powodów osobistych. Jego syn był uszkodzony, jednak nie przez PKU, a u jego kuzynki, urodzonej w 1958 roku, wykryto tę chorobę w 17 miesiącu życia. Dr. Guthrie wynalazł test na początku lat sześćdziesiątych i lobbował za obowiązkowym badaniem na obecność PKU wszystkich noworodków. Jego screening test, wykorzystywany w 24 państwach na świecie, otworzył drogę do badania tą metodą i wykrywania również innych chorób prowadzących do uszkodzenia centralnego układu nerwowego.
NA CZYM POLEGA LECZENIE
Leczenie polega na stosowaniu odpowiedniej diety w oparciu o preparaty niskofenyloalaninowe i bezfenyloalaninowe oraz produkty spożywcze niskobiałkowe. Należy pamiętać, że w terapii fenyloketonurii musimy uwzględniać indywidualną tolerancję fenyloalaniny. Prowadzenie diety wymaga stałej kontroli poziomu tego aminokwasu w surowicy i nadzoru przez lekarzy specjalistów i dietetyków zajmujących się leczeniem tej choroby. Odpowiedzialność za efekty leczenia rozkłada się na działania wielozespołowe: lekarza, dietetyka, psychologa z dużym udziałem samego chorego i jego rodziny. Lekarz prowadzący przede wszystkim ocenia stan zdrowia pacjenta, ustala właściwą, dobrze tolerowaną podaż fenyloalaniny i kontroluje zalecenia dietetyczne. Dietetyk służy fachową poradą w ustalaniu codziennego jadłospisu dla pacjenta. Ważne jest aby porcje żywieniowe spożywane przez pacjentów były właściwie zbilansowane, a posiłki smaczne i spożywane z apetytem. Temu służą również przygotowane przez zespoły specjalistów, przy udziale pacjentów poradniki żywieniowe i broszury z przepisami kulinarnymi. Rygorystyczne utrzymywanie codziennej diety niskofenyloalaninowej jest warunkiem prawidłowego rozwoju dziecka.
Nasze stowarzyszenie dysponuje wieloma opracowaniami poradników wydanymi przy współpracy i pomocy firm MILUPA, SHS International, Mead Johnson i innych.
OŚRODKI LECZENIA
Białystok
Akademia Medyczna Białystok DSK,
Zakład Propedeutyki Pediatrii
Ul. Waszyngtona 17
15-276 Białystok
Tel. 0-85 745 06 24
Bydgoszcz
Akademia Medyczna w Bydgoszczy,
Katedra i Klinika Pediatrii, Alergologii i Gastroenterologii,
Poradnia Gastroenterologii i Alergologii
Ul. Marii Skłodowskiej-Curie 9
85-094 Bydgoszcz
Tel. 0-52 585 42 39
Bydgoszcz
Poradnia Chorób Metabolicznych
Ul. Chodkiewicza 44
85-667 Bydgoszcz
Tel. 0-52 426 21 17
Gdańsk
SPSK 1, Akademia Medyczna w Gdańsku,
Poradnia Chorób Metabolicznych dla Dzieci
Ul.Dębinki
Tel. 0-58 349 28 91
Katowice
Wojewódzki Zespół Ochrony Zdrowia
Matki, Dziecka i Młodzieży,
Poradnia Metaboliczna dla Dzieci
Ul. Powstańców 31
40-038 Katowice
Tel. 0-32 256 19 50
Kielce
Wojewódzki Specjalistyczny Szpital Dziecięcy,
Poradnia Metaboliczna
Ul. Artwińskiego 3a
25-383 Kielce
Tel. 0-41 345 63 25
Kraków
Uniwersytecki Szpital Dziecięcy w Krakowie,
Gabinet Fenyloketonurii,
Poradnia Chorób Metabolicznych
Ul. Wielicka 265
30-663 Kraków
Tel. 0-12 658 20 11 wew. 1236
Lublin
Dziecięcy Szpital Kliniczny im. Antoniego Gębali,
Poradnia Fenyloketonurii,
Ul. Chodźki 2
20-093 Lublin
Łódź
Instytut Centrum Zdrowia Matki Polki,
Poradnia Zaburzeń Metabolicznych
Ul. Rzgowska 281/289
90-261 Łódź
Tel. 0-42 271 17 59
Poznań
Samodzielny Publiczny Szpital Kliniczny nr 5 Akademii Medycznej,
Poradnia Metaboliczna
Ul. Szpitalna 27/33
60-572 Poznań
Szczecin
Pomorska Akademia Medyczna,
II Klinika Pediatrii,
Poradnia Metaboliczna
Ul. Unii Lubelskiej 1
71-252 Szczecin
Warszawa
Instytut Matki i Dziecka,
Poradnia Fenyloketonurii
Ul. Kasprzaka 17a
01-211 Warszawa
Tel. 0-22 327 71 21
Wrocław
Wojewódzki Szpital Specjalistyczny,
Poradnia Metaboliczna
Ul. H. Kamieńskiego 731
51-124 Wrocław
Tel. O-71 327 01 23
Zabrze
Poradnia Leczenia Schorzeń Metabolicznych Szpitala Klinicznego Nr 1
Ul.3 Maja 13/15
41-800 Zabrze
Tel. 0-32 37 04 334
